We use theoretical and atomistic simulation methods, in concert with statistical learning, to understand how fundamental molecular interactions modulate the thermodynamic and kinetic behavior of nanoscale systems, and how these interactions might be manipulated through design of new processes and molecular architectures. Systems we study include polymer and surfactant solution formulations, aqueous solutions and interfaces, and proteins, peptides, and other sequence-specific polymers.
Multiscale modeling techniques
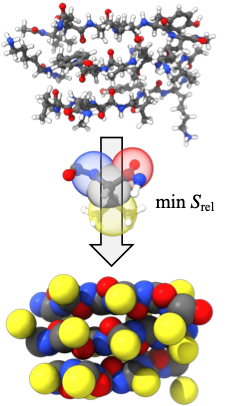
Complex systems with relative entropy coarse-graining
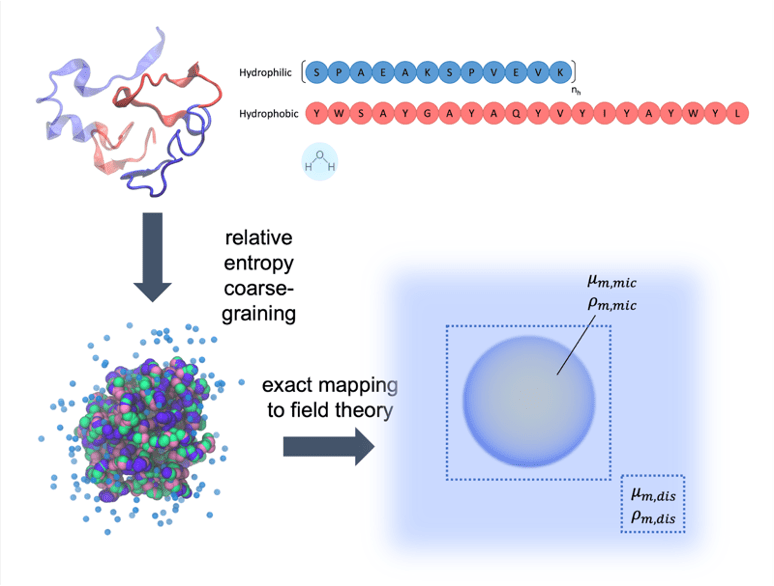
The Shell group pioneered the relative entropy coarse-graining approach, a multiscale modeling technique that enables systematic large-scale simulations of a wide range of complex molecular systems. This approach takes as input small-scale reference all-atom simulations and produces accurate coarse-grained models that can be simulated at much larger scales. Recently developments include a microcanonical coarse-grainingformalism that can be used to create temperature-transferable models by encoding energy fluctuations that would ordinarily be lost during coarse-graining. We have also successfully developed protein folding models using the relative entropy approach that are able to reproduce native structures of globular proteins. Moreover, we have shown that completely bottom-up models parameterized only from atomistic simulations can, without any experimental structural input, predict large-scale amyloid aggregation, and be used to probe the thermodynamics of peptide oligomerization and fibrillization.
Pretti and Shell, Proc. Natl. Acad. Sci. 120, e2309995120 (2023)
Pretti and Shell, J. Chem. Phys. 155, 094102 (2021)
Sanyal, Mittal, and Shell, J. Chem. Phys. 151, 044111 (2019)
Shell, Adv. Chem. Phys. 161, 395 (2016)
Solution formulation using molecularly-informed field theories
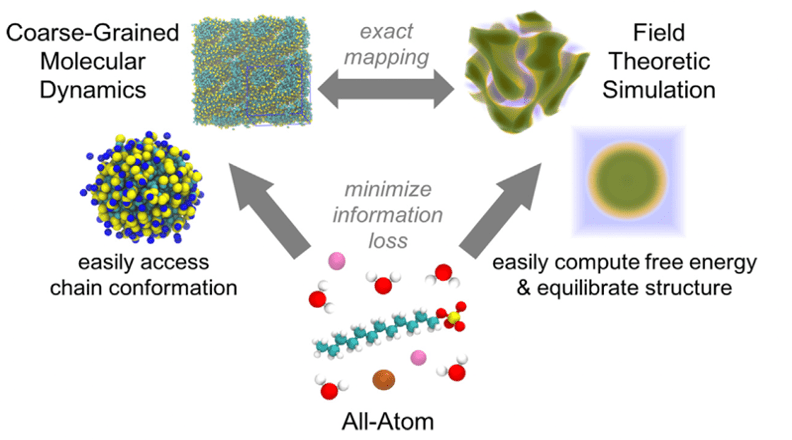
Polymer and surfactant solution formulations are used in a wide variety of technologies spanning consumer products, therapeutics, lubricants, adhesives, paints, and plastics. These multicomponent systems typically show rich solution-phase structures and phase behavior that respond in complex ways to even small changes in chemistry and composition. To model these systems, we are collaborating with the Fredrickson group at UCSB on a multiscale simulation workflow that connects the atomistic to the mesoscale, providing a powerful computational tool for the design of complex formulations involving species of varied molecular weight. Our methodology uses relative entropy coarse-graining to convert representative atomic systems into field-theoretic models; the latter allow direct access to free energies and chemical potentials and rapid equilibration for dense and macromolecular systems. This approach of using molecularly-informed field theory has accurately predicted, without any experimental input, the phase behavior of a range of types of complex formulations, as validated through a variety of experimental measurements.
Nguyen, Dolph, et.al, J. Chem. Phys. 129, 244904 (2023)
Shen, Nguyen, et.al., J. Colloid Interface Sci. 638 (2023)
Nguyen, Sherck, et.al, Macromolecules. 55, 21 (2022)
Sherck, Shen, et.al, ACS Macro. Lett. 10, 576 (2021)
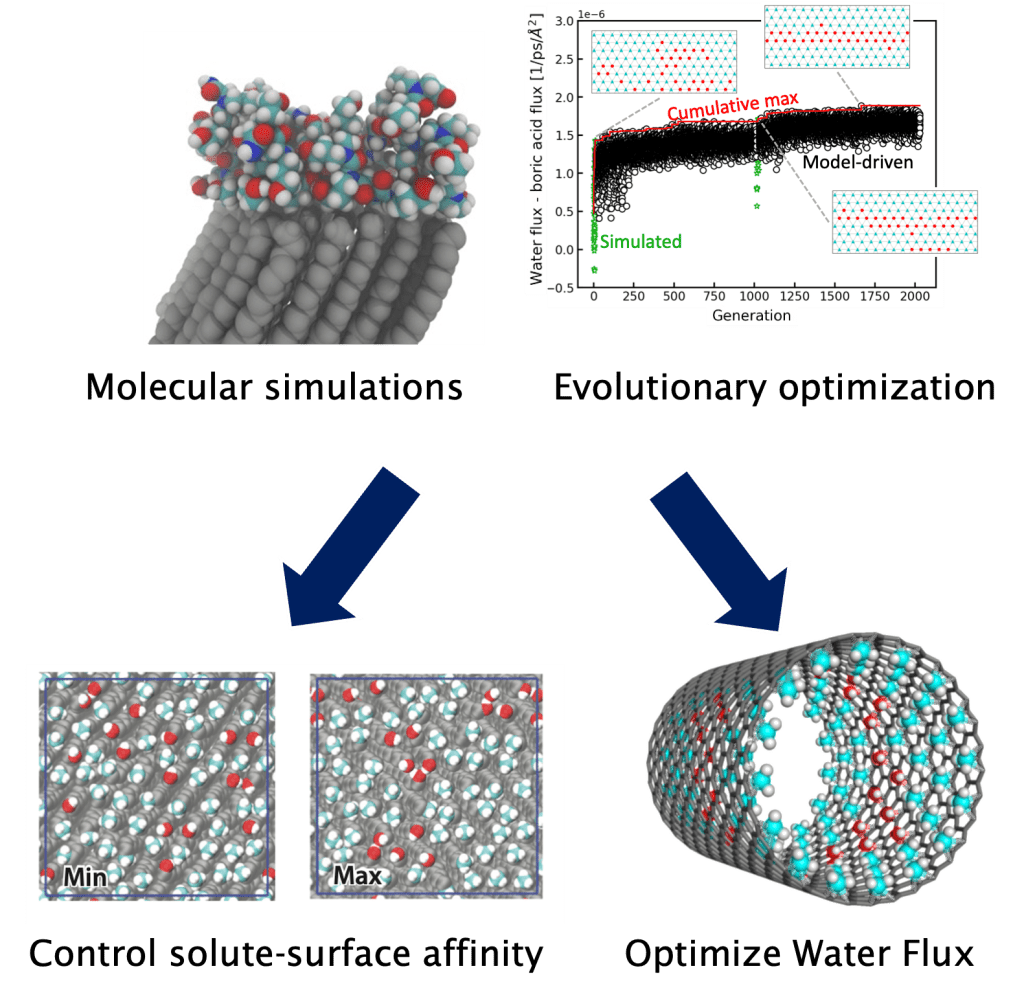
Inverse design of materials
Material synthesis has grown increasingly sophisticated, allowing for the design of materials with precise structures, functional group presentation, and for polymers, monomer sequence. However, the enormous number of possible design solutions requires robust and efficient modeling to understand sequence-structure-property relationships. We are combining advanced molecular simulations with optimization and machine learning workflows to efficiently identify promising materials and elucidate design rules. Currently, these approaches have informed practical materials design for sustainable water treatment, such as the design of membrane pore chemistry and topology for engineering enhanced solute selectivity.
Jiao and Shell, J. Chem. Physical 160, 12 (2024)
Jiao, Katz and Shell, ACS Cent. Sci. 8, 12 (2022)
Monroe and Shell, Proc. Natl. Acad. Sci. 118, e2020205118 (2021)
Biomolecules and bio-inspired molecules
Sequence-defined polypeptoids

Molecular modeling offers direct insight into conformational landscapes, enhancing our understanding of sequence-structure relationships. We are using sequence-specific peptoids (a biomimetic of polypeptides) as a platform for developing design rules for relating chemical sequence to polymer conformation and the dynamics of its hydration waters. Our advanced sampling molecular dynamics methods provide a validated approach for characterizing the conformational landscapes of disordered polypeptoids. Ongoing efforts are investigating peptoids with antifouling properties and developing a bottom-up coarse-grained peptoid model to model and tune peptoid self-assembly and phase behavior.
Jiao*, Rivera Mirabal*, et.al., Biomacromolecules 23, 4 (2022)
Jiao, DeStefano, et.al, Macromolecules 54 (2021)
Understanding fibrillization of tau fragments
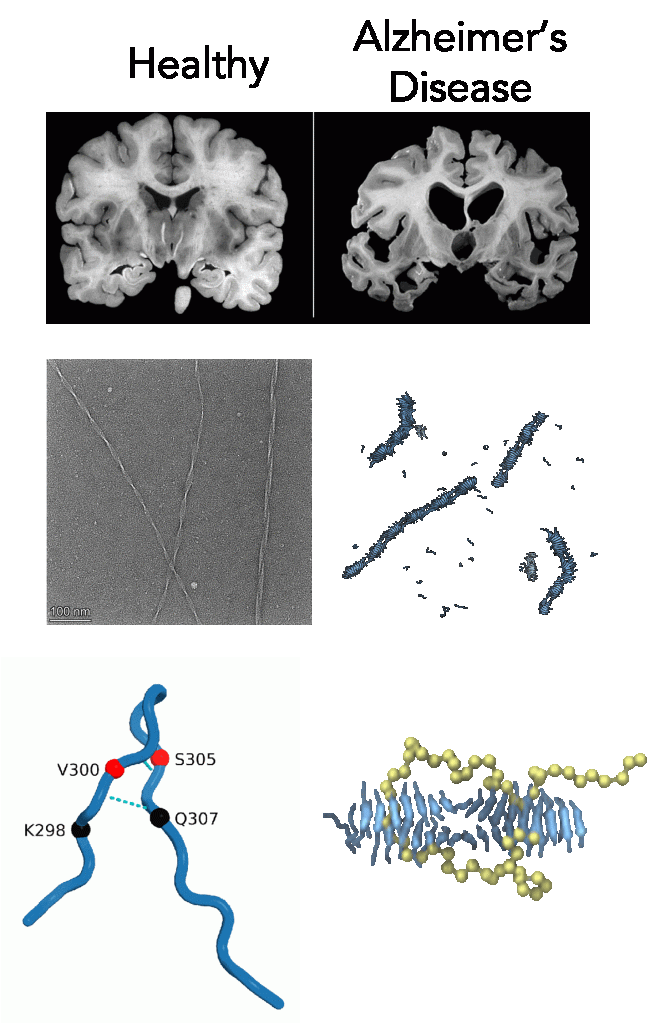
Understanding the molecular mechanisms behind aggregation of the microtubule-associated protein tau is essential for gaining insight into Alzheimer’s and other related neurodegenerative diseases. We are modeling fragments of tau to ask fundamental questions about determinants of tau aggregation. In particular, we have developed a coarse-grained model of a small tau fragment using relative entropy minimization to simulate large-scale fibril formation and investigate the effect of polyelectrolytes like heparin on nucleation. Furthermore, we are studying isoform specific seeding of tau fibrils, uncovering the protective role of intramolecular hydrogen bonds against fibrillization, and the aprotective role of hydrophobic hot spot regions in growing fibrils. These findings offer novel perspectives for tauopathies research and potential therapeutics development.
Pretti and Shell, Proc. Natl. Acad. Sci. 120, e2309995120 (2023)
Longhini, et. al., https://doi.org/10.1101/2023.08.31.555649 (accepted at PNAS)
Vigers, et.al., https://doi.org/10.1101/2023.11.28.568818
pH responsive proteins: Reflectin
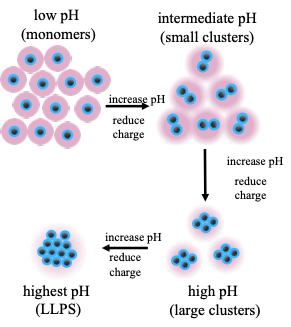
Unlike globular proteins, intrinsically disordered proteins (IDPs) do not adopt a well-defined structure or fold, and can accomplish unique biological functions by modulating their configurations through highly cooperative and dynamic multivalent interactions. As an exceptional example of functional IDPs, the reflectin protein is known for its unique ability to modulate the biophotonic camouflage of cephalopods based on its assembly-induced osmotic properties. Its reversible assembly into discrete, size-controlled clusters and liquid-liquid phase separation is known to depend sensitively on the net protein charge, which makes reflectin a highly stimuli-responsive material and provides an excellent model system to re-engineer a broader class of stimuli-responsive biomolecular materials. In close collaboration with experiments, we are using both atomistic and coarse-grained simulations to gain a fundamental understanding of the relationships between primary sequence, colloidal interactions and assembly, and the macroscopic phase behavior of reflectin. Furthermore, we are exploring ways to engineer minimal reflectin constructs that tailor assembly behavior for targeted bio-synthetic hybrid applications.
Cellulose protein
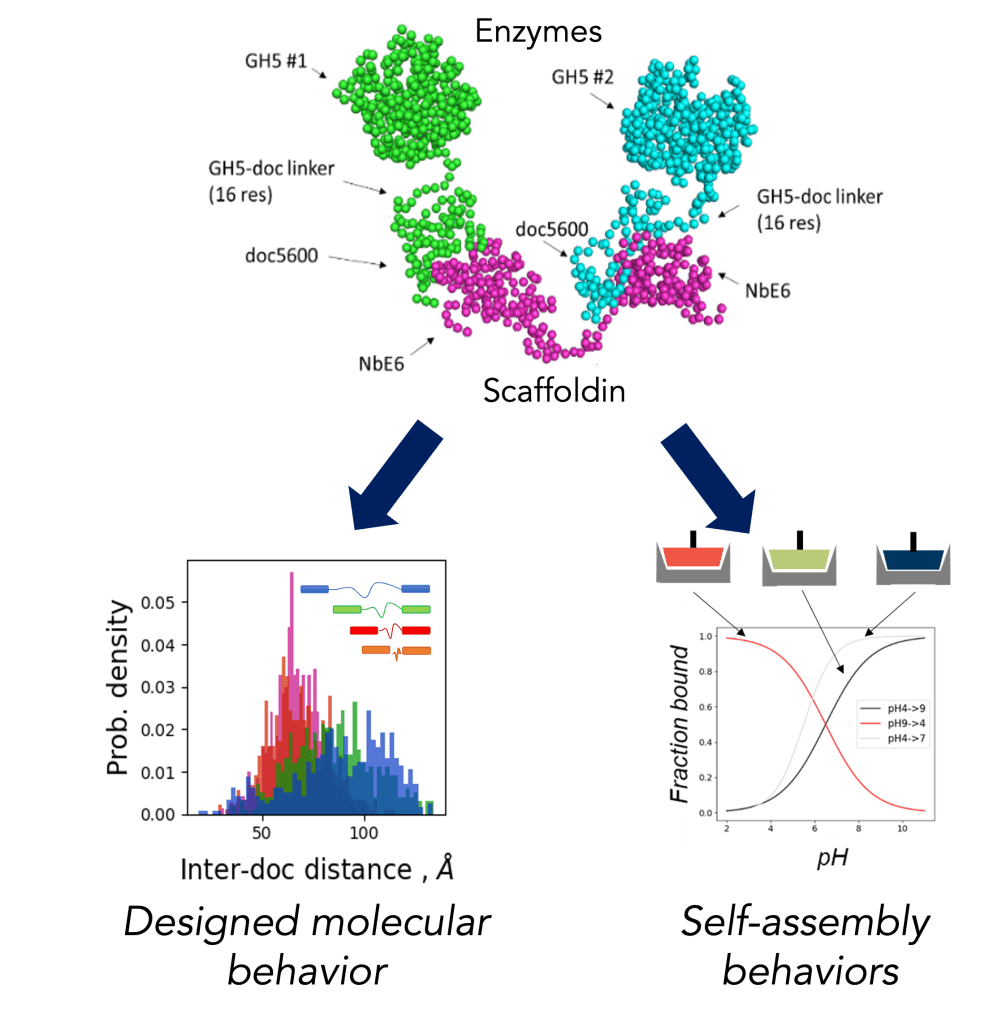
Cellulosomes are multi-enzyme complexes made by anaerobic fungi to degrade plant matter in the guts of herbivores. Using these novel systems as templates, this project is building a synthetic biology platform to create enzyme complexes that rapidly sense-and-respond to their environment, with applications in programmable bioprocessing, bioproduction, and novel biomaterials. In close conjunction with experiments, we are using atomistic and coarse-grained molecular dynamics simulations to both uncover molecular structures and mechanisms of native cellulosome function, and to design synthetic cellulosome complexes that remodel in response to stimuli of interest. Ultimately, we seek to create a framework that responds to a variety of environmental, envisioning a designer protein complex platform that assembles any desired set of proteins in response to many environmental triggers simultaneously.
Complex Formulations
Polymer solutions

Because polymer solutions can adopt a wide range of properties, they find use in an enormous variety of applications such as drug delivery, coatings, lubricants, and personal care products. The versatility of polymer solutions stems from their large design space, where by tuning properties of the polymers, such as their composition, length dispersity, chemistry, and architecture, can dramatically alter properties of the formulations. However, full exploration of this large design space using Edisonian experimental iteration or conventional simulation methods is costly and time-consuming. Using our moleculary-informed field-theoretic methods, we aim to overcome these challenges by enabling fast, ab initio predictions of polymer solution phase behavior with chemical specificity. We have used this approach to predict thermodynamic properties and phase behavior in aqueous solutions with a variety of polymers, polyelectrolytes, ionic and nonionic surfactants, biopolymers, and lubricant additives.
Nguyen, Sherck, et.al, Macromolecules. 55, 21 (2022)
Sherck, Shen, et.al, ACS Macro. Lett. 10, 576 (2021)
Biopolymers as surfactants
With the rising emphasis on sustainability and environmentally friendly practices in industry, there is a growing demand for the development of high-throughput computational methods for screening bio-based molecules as alternatives to conventional petrochemically-derived formulation components in applications such as coatings, consumer products, and drug delivery. Conventional computational tools, such as particle-based molecular dynamics, are commonly used to calculate industrially relevant material properties, including s the critical micelle concentration (CMC). However, such calculations can be extremely computationally demanding or infeasible due to system size limitations. We are leveraging our multiscale modeling approach to show how to enable efficient and accurate predictions of the CMC of a bio-inspired polypeptide surfactant. We have shown that this framework opens possibilities for leveraging molecularly-informed field theories to aid in the study and design sustainable chemistry.
Nguyen, Dolph, et.al, J. Chem. Phys. 129, 244904 (2023)
Aqueous interactions and interfaces
Protein surface hydration
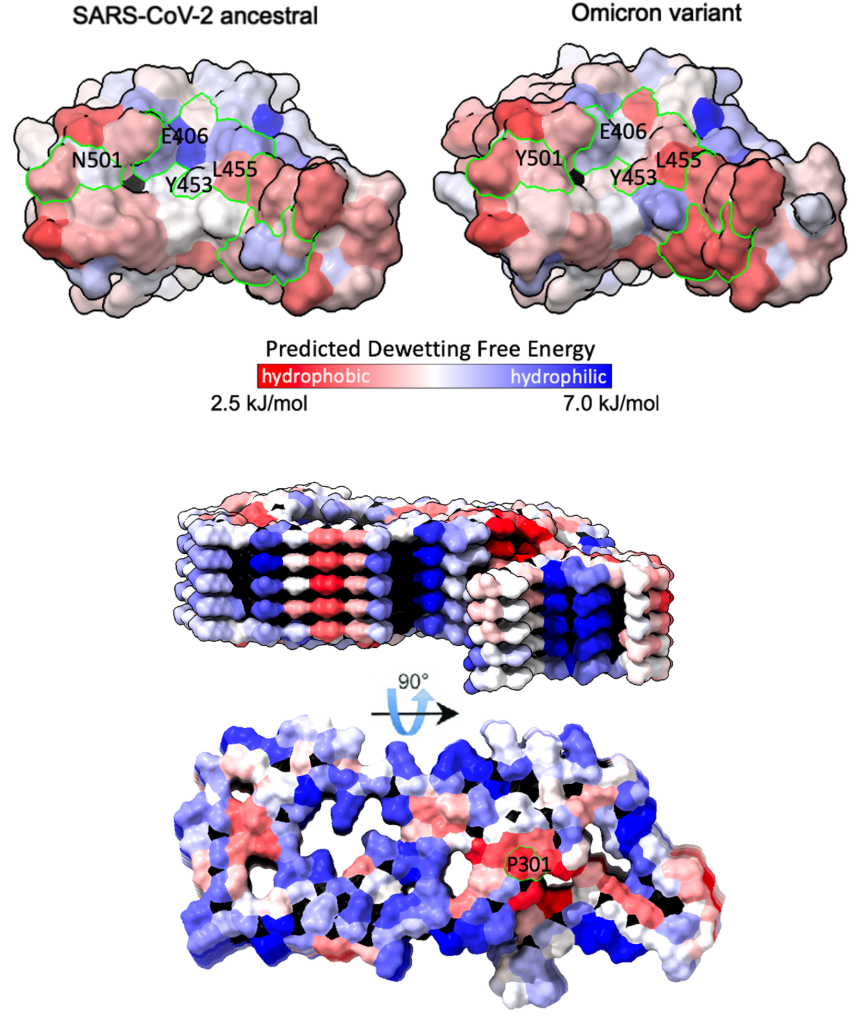
Water plays a crucial role in protein folding, aggregation, solubility, and binding. In particular, accurate modeling of protein-water interactions can uncover complex mechanisms underlying protein stability and binding, and is an exciting new approach towards the development of more effective and targeted therapeutics. Using atomistic molecular dynamics simulations, we are investigating the structure, dynamics, and thermodynamics of water at protein surfaces. These simulations elucidate relationships between surface chemistry, patterning, and hydration behavior, which is essential for e.g. predicting aggregation propensity and protein binding affinity. Furthermore, we use machine learning techniques to efficiently predict dewetting free energies from molecular-scale hydration water structure and energetics.
Najafi, Lobo, Shell and Shea, J Phys Chem B (2025)
Water filtration membranes
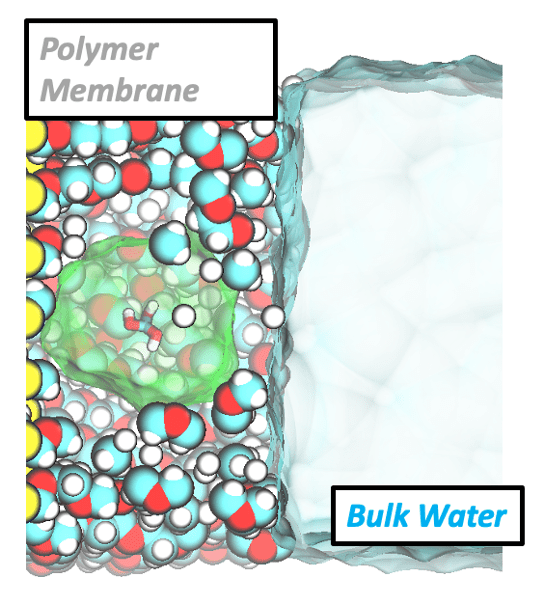
The design of next-generation water filtration membranes requires a fundamental understanding of how water mediates the interactions between foulants/contaminants and complex membrane surfaces. We use atomistic molecular dynamics to investigate the structure, dynamics, and thermodynamics of water and solutes near chemically heterogeneous extended and molecular surfaces. These simulations elucidate mechanisms governing the relationships between surface chemistry, hydration behavior, and solute affinity and selectivity, helping to rationalize and extend previously developed heuristics for membrane design. Using machine learning techniques, we can also elucidate correlations between water structure and dynamics in these hydrated system and develop workflows to predict water structure from experimental measures of water dynamics.
Jiao, Robinson Brown and Shell, Langmuir 40 (2024)
Robinson Brown, et.al, J. Phys. Chem. B 127 (2023)
J. I. Monroe, et.al, Proc. Natl. Acad. Sci. 118 (2021)
